
در وصف اهمیت پروتئین ها همین بس که بدون آنها، حیات به شکل فعلی روی کره زمین، ممکن نبود. بنابراین تعجبی ندارد که جایزه نوبل شیمی ۲۰۲۴ به پژوهشهای پیشگامانه درباره این ماده حیاتی اختصاص یابد؛ پژوهشهایی که بنیان آنها را هوش مصنوعی تشکیل میدهد.
پروتئینها، درشتملکولهایی با عملکرد متنوع و گسترده هستند که واحدهای سازندهی آنها آمینواسیدها میباشند. تعداد، ترتیب (توالی) و پیکربندی فضایی ساختاری آمینواسید ها، نوع عملکرد این درشتملکولها را تعیین میکند. به عنوان مثال پروتئینی را در نظر بگیرید که با قرارگیری روی پایانه های سلولی موجب مختل شدن فرآیند عملکردی آن میشود. توالی آمینواسیدهای سازندهی پروتئینها، برهمکنشهای درونی آنها و نیروهای بین مولکولی مانند نیروی هیدروژنی، موجب شکلگیری ساختارهای منحصربهفرد پروتئینها میشود. این ساختارهای منحصربهفرد منجر به عملکرد اختصاصی این درشتملکولها میگردند.
با این اوصاف، طراحی پروتئینهای کارآمد با عملکردهای خاص، اهمیت بهسزایی دارد. به هر حال طراحیهای آزمایشگاهی و بررسی عملی آنها به کمک روشهای شناسایی مختلفی مثل تشدید روزنانس هستهای و پراش اشعه ایکس، بسیار پرهزینه و زمانبر است، به همین سبب یافتن روشی کارآمدتر با قابلیت تکرارپذیری و صرف هزینه و زمان کمتر اهمیت مییابد.

جایزه نوبل شیمی ۲۰۲۴ که به دیوید بیکر (David Baker) از دانشگاه واشینگتن، دمیس هاسابیس (Demis Hassabis) و جان ام. جامپر (John M. Jumper) از گوگل دیپمایند، اختصاص یافت، دو پژوهش با هدفی مشابه، اما در دو راستای معکوس را دربر میگرفت. بیکر که برنده پنجاه درصد جایزه بود، تحقیقات خود را در مورد طراحی محاسباتی پروتئین انجام داد، بدین صورت که اگر ساختار پروتئینی معینی ارائه شود، میتوان پیشبینی کرد که چه آمینواسیدهایی سازندهی آن هستند.
اما هاسابیس و جامپر که پنجاه درصد دیگر جایزه را به طور مشترک برنده شدند، به پیشبینی ساختار پروتئینی توسط هوش مصنوعی پرداختهاند، بدین صورت که اگر توالی آمینواسیدی ارائه شود، ساختارهای فضایی ممکن پیشبینی میشود. مدل هوش مصنوعی ارائه شده توسط دیپمایند در سال ۲۰۲۱ با نام AlphaFold2 راهحل شگفتانگیز این مسئله بود. معماری این مدل هوش مصنوعی دارای سه فرآیند اصلی شامل جستجوی دیتابیس و پیشپردازش دادهها، ایووفرمر (Evoformer) و ماژول ساختاری است.
در فرآیند اول، دادهها به صورت توالی آمینواسیدی ورودی داده میشود که این ورودی در سه بخش بررسی میگردد: بخش اول، جستجوی عمومی دیتابیس (Generic Database Search) است که خروجی آن ماتریس همراستایی چندگانه توالی (Multiple Sequence Alignment (MSA)) است که توالی آمینواسیدی خاصی را در گونههای مختلف ارائه میدهد. بخش دوم، جفتشدگی (Pairing) آمینواسیدهاست که ارتباط جفتهای آمینواسیدی را نشان میدهد؛ و بخش سوم، جستجوی دیتابیس ساختاری (Structural Database Search) است که قالبهای ساختاری پیشنهادی موجود را ارائه میکند. مجموع اطلاعات بخش دوم و سوم، معرف نمایش جفت (Pair representation) است که مشخصکنندهی ارتباط هر جفت آمینواسید در توالی ساختاری ارائه شده است. خروجی فرآیند اول، نمایش جفتشدگی و نمایش MSA است که هر دو، ورودی فرآیند ایووفرمر هستند. ایووفرمر یک شبکهی عصبی مختص به AlphaFold شامل دو برج عصبی (MSA و Pair) است که باهم ارتباط دارند.
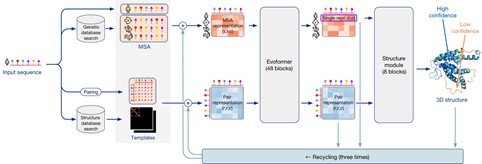
در برج شبکهی عصبی MSA، ورودی ماتریس MSA بررسی میشود. در این بررسی آرایههای سطری مربوط به توالی آمینواسیدهای گونههای مختلف و آرایههای ستونی مربوط به ارزش و شباهت هر آمینواسید در تمام گونهها، ارزیابی و اولویتیابی میشود. در برج عصبی نمایش جفتشدگی، ارزشگذاری هر دو جفت آمینواسید مشابه به عنوان راس (Node) و هر دو جفت آمینواسید غیر مشابه به عنوان لبه (edge) انجام میگردد. بیان راس و لبه بدان جهت است که بررسی هر سه آمینواسید به صورت جفت تشکیلدهندهی مثلثی میشود که به کمک نظریه ناتساوی مثلثاتی ارزیابی ها تکمیل میگردد. تلفیق این دو برج، ماتریس جدید MSA را ارائه میدهد که ارزشگذاری آن اصلاح شده است. این حالت اولین بلوک از ۴۸ بلوک فرآیند ایووفرمر است. خروجی فرآیند ایووفرمر که فرآیندی مشابه الگوریتمهای توجه (Attention algorithms) ارائه شده توسط گوگل است، دو ماتریس نمایشدهندهی MSA و جفتشدگی است. فرآیند آخر، ماژول ساختاری است که ۸ بلوک تکرار شونده است که در هر بلوک وضعیتهای حرکت چرخشی و انتقالی و همچنین محدودیتهای شیمیایی و فیزیکی ساختاری روی اطلاعات ماتریسی توالی حاصل از بخش ایووفرمر اعمال میشود. خروجی این بخش، ساختار سه بعدی اولیه است. برای رسیدن به ساختار سهبعدی نهایی پروتئین، سه بار چرخهی تکرارشوندهی فرآیندی ایووفرمر و ماژول ساختاری انجام میگردد.
در سال ۲۰۲۴ نسخهی جدیدی یعنی AlphaFold3 معرفی شد که نسبت به نسخهی قبلی حدود ۵۰ درصد بهبود یافته بود. این مدل قویتر اما در عین شگفتی سادهتر، نه تنها میتواند برهمکنش پروتئینها را بررسی کند، بلکه توانایی بررسی محاسباتی DNA و RNA و کمپلکس با لیگاندها (کاندیدهای مهم برای طراحی دارو) را نیز فراهم کرده است. امکان بررسیهای متنوع ساختاری، نیازمند آن است تا مدل قدیمی سادهسازی شود و تعمیم الگوریتمهای AlphaFold3 به انواع مختلف محاسبات زیستی امکانپذیر شود. بدین جهت به جای درنظر گرفتن تمامی ویژگیهای ساختاری درشتملکولها اعم از زوایا و چرخشها، تنها محل قرارگیری اتمها در نظر گرفته میشود.
یکی از دلایلی که موجب سادهتر شدن مدل جدید شده، استفاده از الگوریتمهای Diffusion در مدل AlphaFold3 است که عامل کلیدی در ابزار های هوش مصنوعی مانند Midjourney است. اتفاقی که در این مورد رخ میدهد این است که محل قرارگیری تمام اتمها در نظر گرفته میشود و با اضافه کردن نویز به آنها، این اجازه به الگوریتمها داده میشود تا محل درست قرارگیری اتمها را پیدا کنند. این امر باعث میشود تا به جای در نظر گرفتن تمام ویژگیهای ساختارهای موجود، از چند چهارچوب ساختاری که قابل تعمیم به تمام ساختار های موردنظر است، استفاده شود. پس اطلاعاتی که مدل درباره یک پروتئین یاد میگیرد، میتواند برای طراحی ساختار درشتملکولهای دیگر نیز به کار رود و تمام این اطلاعات در یک فضای مشابه، تعبیه (Embedding) میشوند.